Proteomics and proteogenomics analysis of sweetpotato (Ipomoea batatas) leaf and root
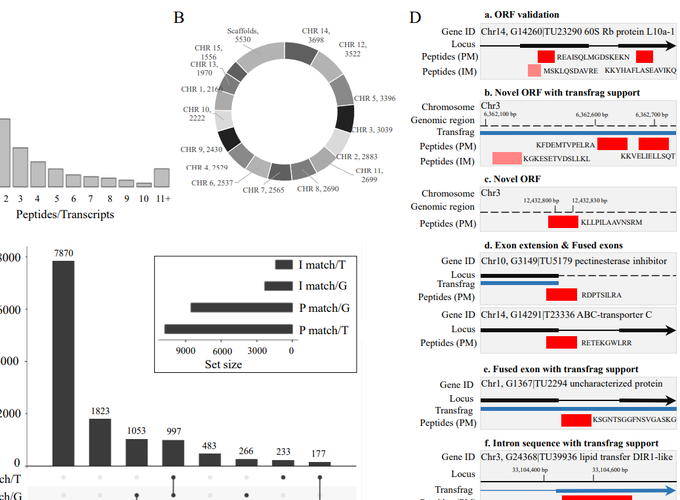 Image credit: Thualfeqar, 2019
Image credit: Thualfeqar, 2019
Proteomics and proteogenomics analysis of sweetpotato (Ipomoea batatas) leaf and root
Abstract
Two complementary protein extraction methodologies coupled with an automated proteomic platform were employed to analyze tissue-specific proteomes and characterize biological and metabolic processes in sweetpotato. A total of 74,255 peptides corresponding to 4,321 nonredundant proteins were successfully identified. Data were compared to predicted protein accessions for Ipomoea species and mapped on the sweetpotato transcriptome and haplotype-resolved genome. The two methodologies exhibited differences in the number and class of the unique proteins extracted. Overall, 39,916 peptides mapped to 3,143 unique proteins in leaves, and 34,339 peptides mapped to 2,928 unique proteins in roots. Primary metabolism and protein translation processes were enriched in leaves, whereas genetic pathways associated with protein folding, transport, sorting, as well as pathways in the primary carbohydrate metabolism were enriched in storage roots. A proteogenomics analysis successfully mapped 90.4% of the total uniquely identified peptides against the sweetpotato transcriptome and genome, predicted 741 new protein-coding genes, and specified 2056 loci where gene annotations can be further improved. The proteogenomics results provide evidence for the translation of new open reading frames (ORFs), alternative ORFs, exon extensions, and intronic ORF sequences. Data are available via ProteomeXchange with identifier PXD012999.